

Absence of evidence of transmission of human idiopathic prions to transgenic models of small ruminants
Most transmissible spongiform encephalopathies diagnosed each year in human patients nowadays (> 80%) are classified as idiopathic prion diseases, that is, without a known cause. In the absence of a better explanation, it is believed that the misfolding of the prion protein, which causes these diseases, occurs spontaneously in the case of idiopathic patients.
In other forms of prion disease, however, the misfolding occurs either due to a mutation in the gene encoding for this protein (genetic prion diseases) or by the entry of an exogenous prion that triggers the misfolding of the prion protein of the patient (acquired prion diseases). The latter group includes iatrogenic cases, caused by medical interventions (such as corneal or dura mater transplantation), cases of occupational disease (by accidental inoculation) or the well-known cases of the variant of Creutzfeldt Jackob disease (vCJD), caused by ingestion of food contaminated with prions of bovine spongiform encephalopathy, also known as the “mad cow” disease.
IRTA-CReSA has been the flagship of the ONE HEATLH approach in the study of diseases for years. This approach means, or wants to emphasize, that pathogens do not function in tight compartments but interact with different animal species, including Homo sapiens, and also with the environment. Therefore, in order to find solutions and get to know the pathogens we deal with, we cannot restrict the study of diseases to a single species.
This is the approach we have taken in the case of the SCRA-ZOO-ECJ project which aims to answer the question: Could it be that the cases of idiopathic Creutzfeldt Jackob disease (CJD) have an origin in prions of small ruminants?
Sheep and goats also have prion diseases: Classical Scrapie, a prion disease acquired and endemic to the Iberian Peninsula, and Atypical Scrapie, an idiopathic (spontaneous) prion disease that is present all over the planet. In principle, no epidemiological evidence has been found to link sheep and goat prions to human prion diseases, and it is now believed that these diseases are not zoonotic. But a few years ago, a group of researchers published experimental results that challenged this hypothesis. Briefly, they inoculated humanized transgenic mice (with human prion protein instead of mouse protein) with classical sheep scrapie prions. As a result, this humanized mouse model was not only infected with sheep prions, but the disease phenotype of those mice was the same as when they became infected with sporadic CJD. This result suggests that sheep and humans may be infected by the same prion strain and pushes us to investigate the subject more thoroughly.
In the SCRA-ZOO-ECJ project we have pursued a similar approach but the other way around. We took prions from 5 human patients from Catalonia and the Basque Country who died of idiopathic prion diseases and inoculated them into transgenic mouse models of small ruminants. Mice with sheep (Tg338 model) and goat (Tg501 model) prions. The aim was to show whether these human prions could actually contain the same prion strain as sheep or goats.
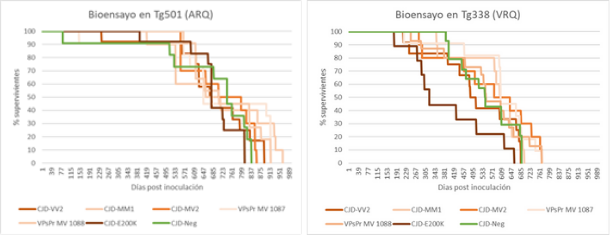
Figure: Survival curves of the two bioassays showing that mice inoculated with human prions had a survival equivalent to those inoculated with a negative control (non-neurological patient brain).
We have reached the end of the 4-year duration of the project and none of the mice we inoculated in the high biological containment unit of IRTA-CReSA (ICTS-RELASB) have become infected on first passage. This is a provisional result that confirms that there is a strong transmission barrier between human and ovine/caprine species. But we can’t rule out the possibility that there has been a subclinical transmission that has gone unnoticed (this is known to happen in this type of experiments). Therefore, it is necessary to carry out according second passages of these bioassays, that are already underway in our facilities, to be able to answer the proposed question. As usual, prion disease research projects take many years to bear fruit, but we are confident that, whatever the outcome, this project will help food policy makers determine whether prion diseases of small ruminants may or may not be a food safety issue.
However, the lack of evidence for a zoonotic risk of Scrapie prions is not evidence of the absence of this risk. It is important, therefore, to continue with programs to monitor and eradicate these diseases.
This project AGL2017-88535-P has been partially funded by the State Research Agency (EAI) of the Ministry of Science and Innovation of the Government of Spain and the FEDER and by the RedPRION project (Interreg POCTEFA EFA148 / 16).














